
Medical Device: U.S. FDA Approval & 510(k)
Partner with KGMP to ensure your products comply with the regulatory standards of your target market.

|
KGMP is a trusted leader in medical devices consulting, providing expert guidance to navigate market-specific requirements and ensure compliance with local and global regulations, facilitating safe export and distribution. |
Medical Device U.S. FDA Compliance: Essential Insights for Your Business
Navigating U.S. FDA medical device approvals requires a thorough understanding of regulatory pathways, including 510(k) premarket notification and PMA (Premarket Approval). Our consulting services provide comprehensive support in preparing regulatory submissions, ensuring compliance with FDA requirements for safety, effectiveness, and quality system regulations (QSR).
KGMP helps demonstrate substantial equivalence for faster market entry, and we offer strategic support on clinical data, risk management, and pre-approval inspections. With evolving FDA regulations, our expertise ensures a smooth approval process.
KGMP Medical Device: U.S. FDA Approval Service

|
|||
|
Device classification and product code |
QMS compliance: CGMP & ISO |
Premarket approval: 510(k) or PMA |
Establishment & Device listing |
Medical Devices Classification and Product Code
The U.S. FDA classifies approximately 1,700 generic types of medical devices, organizing them into 16 panels based on medical specialties. Devices are categorized into three risk-based classes (Class I to III) according to the level of regulatory control needed to ensure their safety and effectiveness.
Each device is assigned a product code based on its classification under 21 CFR Parts 862–892. This product code helps determine whether the device is exempt from 510(k) premarket notification or GMP requirements.
Class I (Low Risk)
- Exempt: No application required, but must comply with general controls
- Non-exempt: 510(k) Premarket Notification required
Class II (Moderate Risk)
- Exempt: No application required, but must comply with general and special controls
- Non-exempt: 510(k) Premarket Notification required
Class III (High Risk)
- All Devices: Premarket Approval (PMA) Application required
QMS (Quality Management System): CGMP & ISO 13485:2016
Manufacturers must implement and maintain Quality Management Systems (QMS) to ensure their products consistently meet regulatory requirements. Current Good Manufacturing Practices (CGMP) requirements for medical devices are at 21 CFR 820.
On January 31, 2024, the FDA issued a final rule amending 21 CFR 820 to align device CGMP requirements with international standards. This amendment incorporates by reference the quality management system requirements of ISO 13485:2016.
510(k) devices are generally reviewed without an on-site inspection, though exceptions may apply. PMA devices require a mandatory pre-approval inspection. Medical device manufacturers registered with the U.S. FDA may be subject to routine GMP inspections.
For more details about KGMP: GMP/ISO compliance service ▶ Click here
Premarket Submission and Approval: 510(k) or PMA
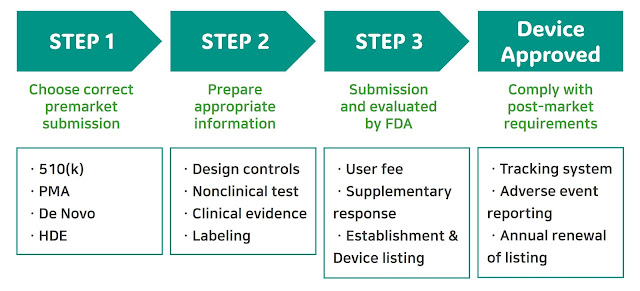
Note: Medical device manufacturers can submit a 513(g) request to the U.S. FDA for clear classification determination.
Q) What is 510(k) Premarket Notification (Evaluating Substantial Equivalence)?
Click to View
A 510(k) is a premarket submission to the FDA to demonstrate that a device is as safe and effective, or substantially equivalent (SE), to a legally marketed device, known as the "predicate." Submitters must compare their device to one or more predicates and support their SE claims.
The predicate must be legally marketed and not in violation of the FD&C Act. Once the FDA determines a device is SE, the device can be marketed in the U.S. The SE determination is typically made within 90 days based on the submitted information.
Types of 510(k) Premarket Notification
- Traditional 510(k): The most common submission, used for devices substantially equivalent to a legally marketed device. It requires detailed information, including device description, intended use, technological characteristics, and performance data. This type requires the most comprehensive documentation.
- Special 510(k): For modified devices that remain substantially equivalent to a previously cleared device. It is a streamlined process for changes in design, material, software, or manufacturing processes that do not significantly affect safety or effectiveness.
- Abbreviated 510(k): Used when a device is substantially equivalent to a predicate and FDA-recognized consensus standards, or guidance documents are available. It allows manufacturers to demonstrate conformity to these standards, reducing the need for extensive testing.
Q) What is PMA (Premarket Approval) Application?
Click to View
A Premarket Approval (PMA) application is the U.S. FDA’s most rigorous regulatory pathway, required for Class III medical devices that pose significant risks. Unlike the 510(k) process, which demonstrates substantial equivalence, PMA requires independent scientific and clinical validation of a device’s safety and effectiveness.
A PMA submission includes comprehensive clinical data, non-clinical performance testing, manufacturing process controls, and risk assessments to substantiate regulatory compliance. The FDA conducts an in-depth scientific review, panel evaluations, and pre-approval inspections of the manufacturing facility to ensure adherence to Quality System Regulations (QSR, 21 CFR Part 820).
Given the complexity and stringent requirements, strategic regulatory planning and expert guidance are essential for a successful PMA approval and long-term post-market compliance.
Q) What is 513(g) “Request for Information”?
Click to View
If a clear equivalent product cannot be identified from existing devices or if it is difficult to find an exact match using the FDA's classification database, a manufacturer can submit a 513(g) Request for Information to the U.S. FDA.
The 513(g) request must include the following information along with a description of the device's characteristics and rationale for its proposed classification:
- Brief medical device description
- Clear intended use
- List or picture of all labeling claims
The FDA provides a recommendation and issues a response letter within 60 calendar days. The letter includes:
- Device determination
- Device class (if applicable, e.g., Class I, II, or III)
- Indication of enforcement discretion, when applicable
- Recommended regulatory pathway (e.g., 510(k), exempt, De Novo, etc.)
- Other relevant information, such as guidance documents, when applicable
Establishment Registration & Device Listing
Owners or operators of establishments involved in the production and distribution of medical devices for use in the U.S., including those imported for export only, must register annually with the FDA and pay the required user fee, with no waivers or reductions for small businesses in FY 2025.
As part of the registration, establishments must list the devices they manufacture and the activities performed on them, a process known as device listing.
All establishments must complete their annual registration between October 1 and December 31 each fiscal year.
For more details about KGMP: Medical device registration and certificate service ▶ Click here
Remember that different regulations apply in each country.
Contact us today to discover how our medical device U.S. FDA approval & 510(k) notification service can benefit your business.
KGMP simplifies compliance with complex medical divice regulations.
K-GMP supports clients in ensuring the quality, safety, and performance of their supply chains while complying with local and global regulations, from ingredients to final product.
 |
 |
 |
|||
 |
 |
 |
|||
 |
 |
 |
The Global KGMP network offers a comprehensive range of one-on-one customized consultancy support for all types of medical device products. We are committed to staying up-to-date with regulatory environments, trends, and industry requirements.
For detailed information about our medical dvice regulation compliance services, please contact our expert directly.

Copyright ⓒ 2012 by KGMP Corp. ALL RIGHTS RESERVED.
© KGMP CORP. and KGMP CORP. RA Consulting, since 2012. Unauthorized use and/or duplication of this material without express and written permission from this site’s author and/or owner is strictly prohibited. Excerpts and links may be used, provided that full and clear credit is given to KGMP CORP. and KGMP CORP. RA Consulting with appropriate and specific direction to the original content.





0 Comments
댓글 쓰기